Partitioning The Taxonomic Tree
by Anthony
28 Nov 2023
Before I started working for Dfam, the entire database was less than a hundred Gigabytes. Just before I joined it doubled in size, and by the time I’d found my feet it was over 800 Gigabytes and growing.

Growth at that rate would strain any system, but a scientific database like Dfam also needs to be downloadable so that researchers can access the data offline, and asking users the world over to download a 800+ Gb file just isn’t reasonable. Even worse, Dfams users are biologists, and likely only interested in one species or group of species to begin with. Asking them to download the entire dataset when they only care about a tiny subset is also not ideal, so I was given the task of coming up with a way of splitting up Dfam into multiple parts in a biologically-relevant way.
The tool used to export and download Dfam was called FamDB, and was written by my predecessor using a python Hierarchical Data Format (HDF) library. I spent some time poking at it before realizing that it needed to be completely refactored from the ground up. The methods that directly stored data in the HDF systems would still work, but the rest of the tool was written with the assumption that all of the data would be in the same file. However, before I started actually rebuilding FamDB, I needed a plan for partitioning the database and handling an unknown number of local files.
After some discussion, we settled on a two-leveled approach. The first, or root file would be mandatory. It would include transposable element (TE) family data for high in the taxonomic tree, as well as the taxonomy data itself. This file would be required with every installation, since it contained the information for searching the tree, but users could add one or more files depending on their area of interest for the actual TE data. After all, there is a significant difference between a search returning “Query Not Found” and “Query Requires File X For A Real Answer,” and the root file allows me to specify to the user which type of failure is occurring. Limiting the file structure to two levels also meant that any user would only have one required root file, and allowed me to not have to account for an arbitrary number of file dependencies.
So how to actually split up the data? Just exporting the data to equally sized files would be trivial, but it was clear that I needed to take the taxonomic tree into account somehow. Here is the algorithm I came up with:
# step 1: build the Taxonomy tree T
# query relevant nodes and all ancestors from the database until a complete tree is formed
relevant_nodes = query the set of all taxonomy nodes with related TE data from the database
parent_nodes = set of the parents of all the nodes in relevant_nodes
while not parent_nodes subset of relevant_nodes:
missing_nodes = query of missing parent_nodes
new_parents = set of parents of missing_nodes
relevant_nodes += missing_nodes
parent_nodes = new_parents
# build T with parent-child relationships and file size for each node
T = {}
for node in relevant_nodes:
T[node.id][parent] = node.parent
T[node.id][children] = node.children
node_sizes = query the approximate file size associated with each node in relevant_nodes
for node in node_sizes:
T[node][size] = node[size]
# recursively calculate the total size for each node + the size of all its descendant nodes
for node in T:
T[node][total_size] = sum_descendant_sizes(T[node])
# assign each node to the root partition
for node in T:
T[node][part] = 0
cache(T)
# step 2: Given a target file Size S, partition T into parts of approximately equal to S, and record in File tree F
F = {}
partition_counter = 1
while T[1][total_size] > S:
# while another file can be partitioned off…
# find the largest node in T still smaller than S
target = -infinity
target_node
for node in T:
if node[total_size] > target AND node[total_size] <= S:
target = node[total_size]
target_node = node
# recursively mark all descendants of target node with partition counter
mark_descendants(node, partition_counter)
# recursively set all descendants total_size to 0
remove_total_size(node)
# recursively subtract the total_size of this partition from all ancestral nodes
subtract_partition(node, target) # save partition data in F
F[partition_counter][filesize] = target
F[partition_counter][root_node] = target_node
partition_counter += 1
# ensure that the root partition is always connected to the root node of each other partition
for file in F:
# recursively mark all ancestors of partition roots as belonging to partition 0
mark_part_0(file[root_node])
# collect all nodes into F for export
for node in T:
partition = T[node][part]
F[partition][nodes] += node
# step 3: export F.json and tree summary and visualizations
...
The basic algorithm is fairly straightforward, essentially finding the largest subtree smaller than the size cutoff and breaking it out of the tree until only a small remainder is left, then tracing a path back from each partition to that remainder. The value of S is tuneable depending on how big we want each file to be, and the actual partitioning step is effectively instantaneous. The slowest part of the process is building and caching the tree and all the filesizes from the database.
The partitioning script has also been extended to weight certain taxa more heavily in case a user wants to append data from another source, or to ensure that some taxons always end up in the root partition, and other smaller optimizations. It also outputs the partitions as a tree in Newick format with a CSV of metadata to be visualized by a follow-on R script. The visualizations look like this:
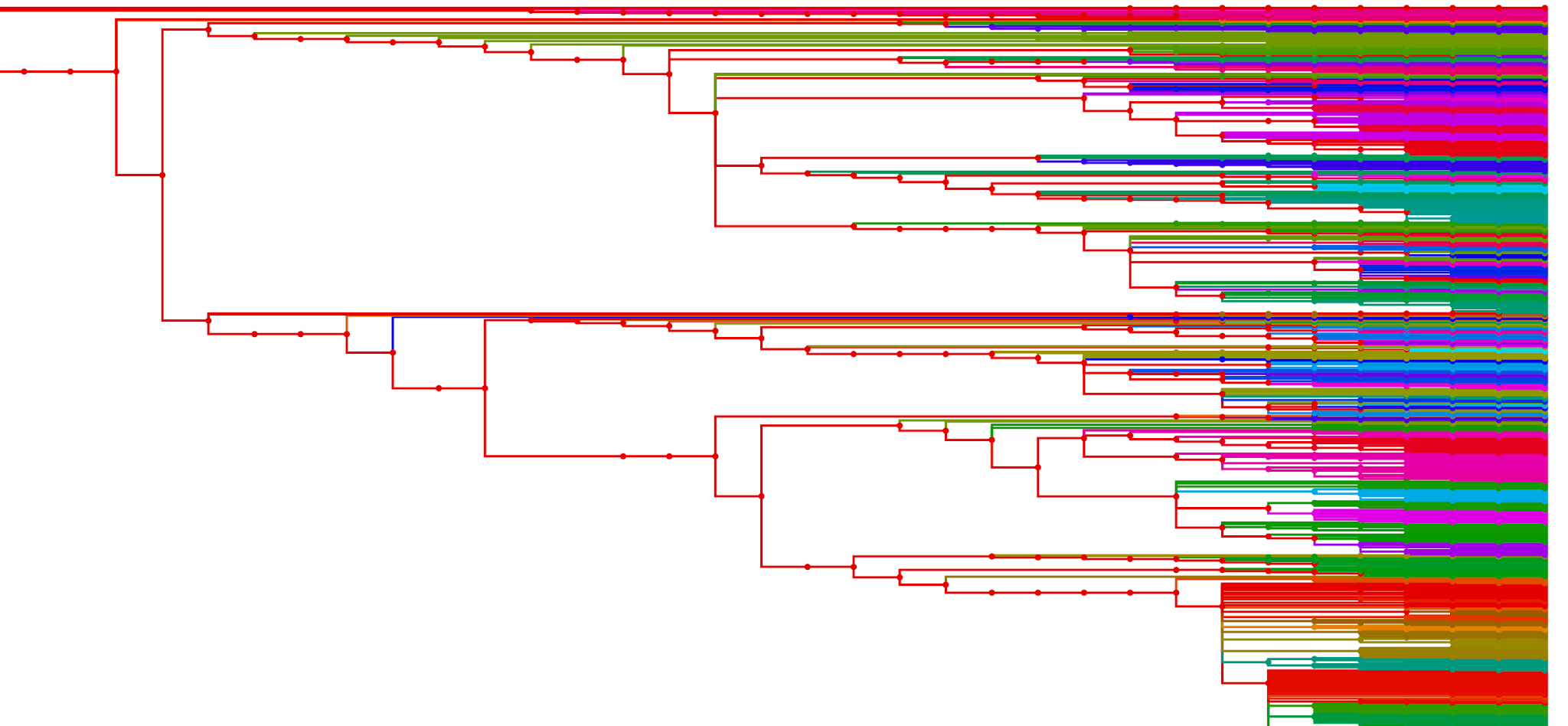
1 Gigabyte Partitions

10 Gigabyte Partitions
There was plenty of other work involved with the re-write of FamDB, as well as all the documentation and set-up needed to make this system work with our Docker container, but I’ll leave those details for another post.